

幽门螺杆菌与上胃肠道疾病
自从澳大利亚的Warren和Marshall[1]于1982年首先从慢性活动性胃炎病人的胃黏膜中分离出幽门螺杆菌(Helicobacter pylori,下称H.pylori)之后, H.pylori与上胃肠道疾病的研究一直是胃肠病工作者的热门课题。目前已经确认H.pylori与上胃肠道疾病中的4种疾病密切相关:①慢性胃炎;②消化性溃疡病;③胃癌;④胃黏膜相关性淋巴组织(mucosa-associated lymphoid tissue, MALT)淋巴瘤(图3-1)。
从H.pylori的发现至今已超过30年的历史,有关H.pylori与上胃肠道疾病之间关系已受到胃肠病学、微生物学、病理学、免疫学及毒理学等领域的学者或专家的极大关注。H.pylori的出现使慢性胃炎和消化性溃疡病面临着一场发病学和治疗学上的革命。关于H.pylori与慢性胃炎及消化性溃疡的关系及其致病机理已越来越明确,而H.pylori与胃癌关系的研究则是热点中的重点。世界卫生组织已将H.pylori列入I类致癌因子[2],因而关于H.pylori与胃癌的研究也备受人们关注, 从基础到临床,从流行病学到致病机理,包括其分子机制的研究,都已在不断深入。然而,要真正揭示H.pylori的致癌机理,许多问题有待进一步深入探索。
(一)幽门螺杆菌的致病因子及致病作用
H.pylori是G-杆菌,呈“S”型或“L”型,长1.5-5.0μm,宽0.3-1.0μm,电镜下可见菌体表面光滑,一端有2-6条带鞘鞭毛,鞭毛顶端膨大呈球形,H.pylori依靠鞭毛运动。因其粘附特性而定植于胃黏膜小凹及其临近表面上皮而繁衍(图3-2,图3-3)。
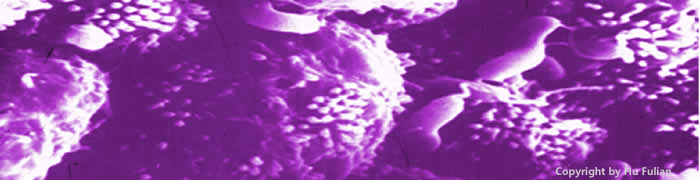
图3-2 H.pylori借鞭毛黏附于胃黏膜上皮细胞表面,此为扫描电镜图象,系从十二指肠溃疡患者胃粘膜中分离出的H.pylori, 可见上皮细胞间沟中有大量H.pylori
图3-3 此为胃粘膜上皮细胞表面的H.pylori以及由H.pylori引起的胃粘膜炎症反应, 可见上皮细胞下面有大量中性粒细胞皮浸润。
H.pylori的致病机理非常复杂,H.pylori致病因子对胃黏膜的损伤及其对人体损伤机制至今尚未完全明了。目前认为H.pylori的致病机制包括:H.pylori的定植、毒素引起的胃黏膜损害、宿主的免疫应答介导的胃黏膜损伤以及H.pylori感染后胃泌素和生长抑素调节失衡所致的胃酸分泌异常等。参与H.pylori致病的因子分为定植因子和毒力因子等。其中定植因子是H.pylori 感染的首要条件。H.pylori 本身的动力装置、黏附特性、具有毒性作用的酶以及多种毒素既有利于其定植,也有助于H.pylori在高酸环境下存活,最终是否致病,有赖于H.pylori菌株的不同及宿主的差异以及环境因素的影响。
H.pylori致病因子很多,按其致病机理及其特点,通常将H.pylori 致病因子大致分成四大类:①与H.pylori定植有关的致病因子;②以损伤胃黏膜为主的致病因子;③与炎症和免疫损伤有关的致病因子;④其它致病因子。在诸多的H.pylori致病因子中,其中以H.pylori产生的尿素酶在H.pylori的致病机理中起十分重要的作用。H.pylori能水解尿素释放出氨(NH3),直接对胃黏膜造成损伤,而H.pylori本身在其产生的“氨云”包绕之中则免受胃酸、胃蛋白酶的侵袭,使其在很低的pH环境中得以生存。H.pylori产生的分子量为87kD的空泡细胞毒素(vaculating cytotoxin A, VacA)以及分子量为128kD的细胞毒素相关蛋白(cytotoxin-associated protein, CagA)是H.pylori的重要致病因子。感染了Tox+/CagA+ H.pylori菌株病人的胃窦黏膜中有大量中性粒细胞浸润,其机理可能与通过增加胃黏膜上皮分泌白细胞介素-8(interleukin-8,IL-8)有关。H.pylori毒素与H.pylori的其它致病因子如脂多糖、蛋白酶、脂酶、磷脂酶A2等共同作用,对胃黏膜产生局部的炎症反应和免疫反应,使胃黏膜遭受炎症和免疫损伤,而损害的胃黏膜则更容易遭受胃酸、胃蛋白酶的侵袭。H.pylori可以引起浅表性胃炎,H.pylori持续感染可以从浅表性胃炎发展成萎缩性胃炎、肠上皮化生和非典型增生,而萎缩性胃炎、肠上皮化生和非典型增生(黏膜内瘤变),都是属于癌前期病变。现已认为重度H.pylori相关性胃炎与非贲门部胃腺癌密切相关,世界卫生组织已将H.pylori定为Ⅰ号致癌因子。因此可以说H.pylori是胃癌的始动因子。
全球感染H.pylori的人超过50%,然而大多数人不发病,只有少数人发展为不同的临床疾病,这是什么原因?可能由以下因素决定:(1)个体差异;(2)菌群差异;(3)环境差异;(4)处在H.pylori感染过程中的不同阶段。
关于H.pylori感染与上胃肠道疾病的关系以及可能发生的不同临床疾病可参考以下模式图(图3-4):
(二)幽门螺杆菌与上胃肠道疾病的关系
1.幽门螺杆菌感染与慢性胃炎
慢性胃炎病人中,H. pylorip感染率超过95%,其感染率随着年龄增加而增加[7]。Hp感染可以引起三种不同类型胃炎[3]:①浅表性胃炎(superfecial gastritis);②弥漫性胃窦炎(diffuse antral gastritis);③多灶性萎缩性胃炎(multifocal atrophic gastritis)。H. pylori相关性胃炎的病理特点是:①黏膜上皮变性;②中性粒细胞和慢性炎症细胞浸润和/或显著的淋巴滤泡形成;③肠上皮化生;④非典型增生(黏膜内瘤变);⑤腺体萎缩。
慢性胃炎病理诊断标准按"直观模拟评分法(visual analogue scale)" [4] 包括5项组织学变化和 4个分级:5项组织学变化包括H. pylori感染、慢性炎性反应(单个核细胞浸润)、活动性(中性粒细胞浸润)、萎缩(固有腺体减少)、肠化(肠上皮化生); 4个分级包括0提示无,+提示轻度,++提示中度,+++提示重度。
上皮退行性改变是指黏液耗损、上皮细胞变性、渗出及脱落等均是慢性胃炎的显著特征。老年性慢性胃炎的特点是肠上皮化生和腺体萎缩的发生率增高,随着腺体的消失也可出现糜烂或溃疡形成,腺体萎缩可能是细菌作用的结果,也可能是长期慢性炎症的反应。H. pylori感染引起的肠上皮化生是胃肠道黏膜对慢行持续性感染的一种适应现象。根据黏液含量和细胞形态可将肠上皮化生分为3种主要类型:①Ⅰ型(完全型):化生上皮与正常的小肠型上皮相似;②Ⅱa型(不完全型)③Ⅱb型或Ⅲ型(不完全型):其柱状上皮与分泌硫酸黏液的结肠上皮相似,Ⅲ型肠化是发展为胃腺癌的高危因素,随着肠化生的加重,不适合H. pylori的定居,因而细菌逐渐消失,H. pylori的消失则伴随着慢性胃炎的后期H. pylori检出率降低或消失,伴随着慢性炎症细胞的减少或消失。
H. pylori持续感染,可以从浅表性胃炎发展成萎缩性胃炎,肠上皮化生和非典型增生(黏膜内瘤变)。而萎缩性胃炎、肠上皮化生和非典型增生(黏膜内瘤变),都属于癌前病变。现已认为重度H. pylori相关性胃炎与非贲门部胃腺癌密切相关。H. pylori是慢性活动性胃炎的重要病因,其证据符合Koch法则,即病原体存在于患者体内,其存在部位与病变部位一致,清除病原体后病变好转,该病原体在动物体内可诱发与人相似的疾病。
2.幽门螺杆菌感染与胃癌
1994年世界卫生组织下属的国际癌肿研究机构将H. pylori列入胃癌的I类致癌因子,这是根据流行病学资料以及对胃癌发生过程中演变规律的认识所取得的共识。
流行病学方面支持H. pylori感染致胃癌的主要论据为:①H. pylori感染率与胃癌发生率呈明显正相关,感染者比非感染者患胃癌的风险值增加(有地区差异,如非洲就例外);②H. pylori感染与胃癌的发生都随着年龄的增加而增加;③H. pylori主要定居于胃窦,与胃癌的好发部位一致。
流行病学调查研究表明:胃癌高发区,也是H.pylori感染高发区,而且感染的年龄很早。有调查资料表明:胃癌死亡率由低到高的地区,H.pylori感染率亦由63%上升致96%。H.pylori感染者其胃癌发生的风险值较非感染者高。国内有一项大的前瞻性研究调查了18244名自然人群,随访10年,H.pylori阳性者较H.pylori阴性者胃癌发生率高,OR值为1.84。然而亦有一些流行病学调查却显示不同的结果,即胃癌的发病率与H.pylori感染无明显的关系。
流行病学的调查只是反应H.pylori与胃癌发生的相关比,尚无证据证明H.pylori感染如何引起胃癌的发生。H.pylori本身并不分泌致癌物,它导致胃癌的发生是一种间接的形式,如H.pylori所含的空泡毒素、尿素酶等毒力因子可损伤胃黏膜细胞,造成黏液排空,上皮脱落,电镜下可见胃黏膜细胞肿胀,细胞内质网系统扩张。H.pylori引起炎症反应并释放炎性介质,致使细胞增殖加快,增生活跃的细胞DNA合成旺盛,易受基因毒致癌物的损伤而发生细胞突变、缺失,而导致细胞癌变。H.pylori感染首先引起胃黏膜的炎症改变,长期的慢性炎症将导致胃黏膜向胃癌方向演化[5]。Correa[6]描述了肠型胃癌发生的自然病史,由正常胃黏膜→浅表性胃炎→萎缩性胃炎→肠上皮化生→非典型增生→胃癌。H.pylori感染与肠型胃癌和弥漫性胃癌都有关,但一般认为与肠型胃癌关系更为密切。但这是一个漫长的过程,H.pylori只是作为许多致癌因子之一而作用于这一过程的某一阶段。许多研究资料显示,在H.pylori高流行地区H.pylori感染者较未感染者肠化生率为高(43%与25%),与胃癌关系最密切的Ⅲ型肠化发生率在H.pylori感染高流行区(28%)明显高于H.pylori感染低流行区(17%)。H.pylori主要集聚在胃窦,也是肠化生和异型增生以及胃癌发生率最高的部位。可以认为,H.pylori感染是肠化及异型增生的重要因素,早期感染H.pylori可以导致并加速肠化生及异型增生的发生,促使正常胃黏膜向胃癌方向演化。国内外都有研究报道,在H.pylori根除之后,部分肠化生和异型增生可以逆转。如果H.pylori感染持续存在,则H.pylori感染对胃黏膜造成的损伤可以改变H.pylori本身的生存环境,虽然在相当一部分胃黏膜肠化的早期阶段可以检出H.pylori,但随着病变的加重,H.pylori不能适应环境的改变而最终消亡,这就是人们认为H.pylori不能定居在肠化生部位的原因。
H.pylori感染时可以引起胃癌相关基因的变异,包括原癌基因如ras,c-met,c-myc,c-erbB-2等原癌基因的激活;而抑癌基因p53突变、失活。我们的研究发现在癌前期病变中H.pylori感染者c-met基因表达率(61.4%)明显高于未感染者(35.4%),在浅表性胃炎、萎缩性胃炎、肠化生、及非典型增生病变中,c-met的表达率和过表达率分别为22.2%(5.5%);44.1%(26.4%);67.6%(37.8%);61.9%(38.1%);在胃癌组为69.2%,随着病变的加重,从浅表→萎缩→肠化→非典型增生→胃癌,c-met表达及过表达率逐渐增加[7]。在体外,利用H.pylori培养滤液与GES-1细胞一起培养,可以引起GES-1细胞c-met、c-myc原癌基因的mRNA 的过表达,表明毒素对GES1细胞的生长分化有一定的影响[8]。
Parsonnet提出H.pylori导致胃癌的三种假说[9]:①细胞的代谢产物直接转化胃黏膜;②类似病毒的致病机理,H.pylori的DNA整合到宿主胃黏膜细胞中,引起转化;③H.pylori引起炎症反应,而炎症有基因毒作用,破坏DNA导致基因突变和恶性转化。以上的研究大都支持第三种学说。其研究结果表明与H.pylori引起的炎症有关。有报道将H.pylori感染蒙古沙鼠于1-1.5年之后成功的诱发胃癌,而且是经过了炎症细胞浸润→萎缩性胃炎→肠上皮化生→非典型增生→胃癌的演化过程[10]。目前也有人试图将H.pylori-DNA整合到胃黏膜细胞染色体中,以此来阐明H.pylori致胃癌的机理,但至今尚未见到成功的报道。近期有关胃癌发生的一个潜在的重要发现是胃癌细胞的起源可能不是来源于胃上皮细胞本身,而是骨髓起源的细胞在H.pylori的存在和作用下分化来的胃上皮细胞[11],如果这观察被证明属实,它将从本质上影响和改变H.pylori 相关胃癌的治疗以及和慢性炎症有关的其它上皮癌症的治疗。关于H.pylori如何引起胃黏膜转化,包括对细胞膜、细胞质的传导、以及对DNA的合成转录等方面的直接或间接影响,都有待今后作更多更深入的研究。
H. pylori致胃癌的发生是一个漫长的过程:H. pylor感染首先引起胃黏膜的炎症改变,长期的慢性炎症将导致胃黏膜向胃癌方向演化。从炎症开始,经若干癌前病变的中间阶段,最后发生癌变。H. pylori感染与肠型胃癌和弥漫性胃癌都相关,但与肠型胃癌关系更密切,H. pylori只是作为许多致癌因子之一而作用于这一过程的某一阶段。H. pylori致胃癌的发生是一个漫长的过程:H. pylor感染首先引起胃黏膜的炎症改变,长期的慢性炎症将导致胃黏膜向胃癌方向演化。从炎症开始,经若干癌前病变的中间阶段,最后发生癌变。
3.幽门螺杆菌与消化性溃疡
(1)幽门螺杆菌的发现是消化性溃疡在发病机理及病因学上的革命
消化性溃疡的发病机理非常复杂,通常认为溃疡的发生是因为损害因素与防卫因素之间的失衡,损害因素是包括胃酸、胃蛋白酶、幽门螺杆菌、非甾体类消炎药、酒精、吸烟、胆汁反流及炎性介质等;防御因素包括胃黏膜-黏液屏障、重碳酸盐、磷脂、黏膜血流、细胞更新、前列腺素和表皮生长因子等。在攻击因子中胃酸起着主导作用。早在1910年Schwartz的名言“没有胃酸就没有溃疡”,所以胃酸一直在消化性溃疡病的发病机理中占据统治地位。自从1982年Warren和Marshall从慢性活动性胃炎病人的胃黏膜中分离出H.pylori之后,H.pylori在溃疡病发病机理中的作用对胃酸形成挑战,有些学者也提出“没有H.pylori就没有溃疡”;“没有H.pylori就没有溃疡复发”。随着人们对溃疡病发病机理的新认识,自然对溃疡病的治疗策略亦有新的变更, H.pylori的发现使消化性溃疡在发病学和治疗学上面临着一场革命。Schwartz的名言“没有胃酸就没有溃疡”至今沿用不衰,所以针对抑制胃酸分泌的药物始终是治疗溃疡病的主要手段, 但当今新观点还必须加上“没有H.pylori就没有溃疡和溃疡复发” 。关于H.pylori相关性溃疡如果不根除H.pylori则停用抑酸药后溃疡就会复发,必须根除H.pylori之后才能降低或预防溃疡复发这一事实已被大家普遍认可。消化性溃疡发病非常复杂,从整体上讲,大约有5-10%的消化性溃疡并没有合并H.pylori感染, 这些溃疡可能与长期服用阿司匹林/NSAIDs等药物而使胃黏膜屏障遭受破坏有关。所以当今溃疡病的治疗原则是在传统的抑酸治疗的同时, 必须根除H.pylori和保护胃黏膜。现在充分的理论依据证明了H.pylori的发现使溃疡病的发病机理和治疗策略发生了新的变更。
(2)幽门螺杆菌在消化性溃疡形成中的致病作用及其致病机理
1)幽门螺杆菌与消化性溃疡复发的关系:“愈合” 与“治愈” 是两个概念不相同的医学术语,在H.pylori未发现之前,消化性溃疡被认为是原因不明的复发性疾病, 通常认为消化性溃疡只能“愈合”, 而不能“治愈” ,应用抑酸药或者维持治疗都只是使溃疡暂时愈合,但一旦停止治疗则溃疡很快复发。因此以往的观点认为消化性溃疡是一个不可治愈的疾病。自从1982年发现H.pylori后,对于消化性溃疡的自然病程有了新的认识,国内外大量临床研究证实在根除H.pylori后可以降低或防止胃及十二指肠溃疡的复发。Mohamed[12]集成分析700例十二指肠溃疡患者的复发情况,H.pylori未根除患者1年内溃疡的复发率为80%,而H.pylori根除患者复发率仅为4%,胃溃疡亦是如此。我们过去的一组研究亦证实H.pylori根除者溃疡完全愈合,未根除者其愈合率61.9%。随访半年,H.pylori根除者半年内无复发,一年内复发率4%,H.pylori未根除者半年内复发率58%,一年内100%复发[13]。北京地区有一项对248例十二指肠溃疡患者作H.pylori根除治疗随访一年的多中心的临床研究,其研究结果表明,H.pylori根除组溃疡复发率仅2.3%,而在H.pylori未根除组,一年复发率58.9%[12]。20多年来对H.pylori相关性溃疡的治疗研究证实,消化性溃疡是一个可以治愈的疾病。
2)幽门螺杆菌在溃疡形成中的致病机理:幽门螺杆菌致胃十二指肠黏膜损伤的机理十分复杂,目前主要以下4种学说:
①“漏屋顶学说”:Goodwin[14]把发炎的胃黏膜比喻为漏雨的屋顶,无雨则暂时的干燥,意思是说无胃酸就无溃疡。在给予抗分泌药之后,胃酸抑制,溃疡愈合,但只能获得短期的疗效,因为终究没有把漏雨的屋顶修好,没有改变溃疡病的自然病程。消化性溃疡的自然病程中溃疡复发率>70%。如果针对与炎症及与溃疡有关的H.pylori治疗(根除H.pylori),则溃疡不易复发。所以只有通过黏膜修复即修好屋顶才能长期防雨,即达到溃疡病治愈的目的。
②“胃泌素相关学说”:Levi[15]提出H.pylori周围的氨云可使胃窦部pH值增高,胃窦部胃泌素反馈性释放增加,因而胃酸分泌增加,在十二指肠溃疡的形成中起重要作用。对于H.pylori相关性十二指肠溃疡,如果能够真正根除H.pylori,溃疡是不应该复发的,再感然的发生率很低,西方国家大约每年1%左右。
③胃上皮化生学说[16]: H.pylori通过定植于十二指肠内的胃化生上皮,引起粘摸损伤并导致十二指肠溃疡形成。H.pylori释放的毒素及其激发的免疫反应导致十二指肠炎症的产生。由于炎症黏膜对其他致溃疡因子的攻击耐受力下降,导致溃疡的发生,或者重度炎症本身导致溃疡产生。在十二指肠内,H.pylori仅在胃上皮化生部位附着定植,此为本学说的一个有力证据。
④介质冲洗学说:已经证实H.pylori感染导致多种炎性介质的释放,这些炎性介质在胃排空时冲至十二指肠而导致十二指肠黏膜损伤。加上H.pylori可以定植于有胃上皮化生的十二指肠黏膜,这就解释了H.pylori主要存在在胃窦但可以导致十二指肠溃疡的发生。
3)幽门螺杆菌与难治性溃疡的关系:应用H2RAs治疗,十二指肠溃疡治疗8周;胃溃疡治疗12周,若溃疡仍未愈合,一般认为属于顽固性溃疡。H.pylori感染与NSAIDs的应用可能为顽固性溃疡的重要潜在因素,大量吸烟、酗酒以及胃酸分泌量过多(如胃泌素瘤)等因素均可使溃疡延迟不愈。
持续的H.pylori感染是顽固性溃疡的一个重要因素,许多研究资料表明根除H.pylori可以加速顽固性溃疡的愈合和降低高复发率。我们曾发现6例经H2RAS持续治疗半年而溃疡未愈合的十二指肠溃疡病人,经检查全部合并H.pylori感染,但经抗H.pylori感染治疗之后,其中5例溃疡愈合,另外1例溃疡明显缩小[17]。所以对顽固性溃疡应仔细检查H.pylori,对于合并H.pylori感染的顽固性溃疡应进行H.pylori根除治疗。对于H.pylori阴性的顽固性溃疡则应针对其它影响溃疡愈合的因素进行处理。
4)幽门螺杆菌阳性的消化性溃疡治疗新策略:现在有充分的理论依据证明了H.pylori的发现使得消化性溃疡的发病机理产生了重大变更, 所以随着消化性溃疡发病机理的改变, 其治疗策略亦发生了重大变更。当今消化性溃疡的治疗策略应该包括三个方面:①抑制胃酸;②根除H.pylori ;③保护胃黏膜。沿用这三条原则才能达到治愈溃疡的目的。
4.幽门螺杆菌与胃MALT淋巴瘤
早在1983年就有学者提出胃肠道淋巴组织学特点和临床生物学特点的建议,因为这个建议是针对胃黏膜相关淋巴样组织(MALT)淋巴瘤,而非结节样淋巴组织淋巴瘤而提出的[18],这类淋巴瘤起源于结外边缘带,黏膜相关淋巴组织(如胃、唾液腺等),胃MALT淋巴瘤由于通过胃镜检查而易于取材,成为目前研究热点。
正常胃黏膜是缺少淋巴组织的,感染H.pylori之后,胃黏膜组织中有淋巴滤泡形成,进而MALT型淋巴样组织在胃内聚积[19, 20],所以这种淋巴瘤是“获得性MALT”,本病无特异的临床症状,内镜下显示胃黏膜充血或糜烂,少见有肿瘤增生样改变。组织病理学特点为:结外淋巴瘤,由形态不同的小B细胞构成,包括边缘区(中心细胞样)细胞、单核细胞样细胞、小淋巴细胞和散在免疫母细胞以及中心母细胞样细胞。部分病例存在浆细胞样分化。肿瘤长生长在边缘区,围绕反应性B细胞滤泡,进一步扩展到滤泡内区域,肿瘤细胞浸润胃腺体的上皮为其病理特点,形成淋巴上皮病变[21]。
胃MALT淋巴瘤的治疗:早期根除H.pylori胃MALT淋巴瘤可以缩小或消失,Wotherspoon等[22]应用抗生素根除H.pylori后使胃MALT淋巴瘤消退,为胃MALT淋巴瘤的致病机理的探讨及治疗的研究提供了可靠的依据, 并不断地被许多学者的研究所证实[23]。胃MALT淋巴瘤的预后是比较好的,然而,对抗生素治疗的反应因MALT浸润黏膜的层次不同,其肿瘤消退率截然不同。对于浸入黏膜下层、肌层、浆膜层或远隔脏器转移者,对抗生素无反应,对存在t(11:18)易位的患者预后亦差。但从原则而言,凡是H.pylori阳性的MALT淋巴瘤一律应该做根除治疗。
H.pylori相关性疾病的治疗参照最新的国内外共识处理意見[25-26],有关治疗的具体方案参照本书的有关章节,但具体治疗时则强调个体化治疗。
参考文献
1. 胡伏莲. 重视幽门螺杆菌与上胃肠道疾病关系的研究. 中华医学杂志, 1998,78(7):483-484.
2. International agency for research on cancer. Schitosomes, live flukes and helicobacter pylori. IARC monographs on the evaluation on carcinogenic risks to humans. Vol61, Lyon: IARC, 1994.
3. 沈祖尧,梁伟强. 幽门螺杆菌与胃炎. 见胡伏莲,周殿元. 幽门螺杆菌感染的基础与临床. 北京:中国科学技术出版社,2002:147-152.
4. 中华医学会消化病学分会.中国慢性胃炎共识意見.(2012年,上海).中华消化杂志2013,33(1):16
5. Crowe SE. Helicobacter infection, chronic inflammation and the development of malignancy. Curr Opin Gastroenterol, 2005, 21: 32-38.
6. Correa P. A human model of gastric carcinogenesis. Cancer Res, 1988, 48: 3554-3560.
7. 郭飞,胡伏莲,贾博琦。幽门螺杆菌感染者胃黏膜癌前病变与c-met原癌基因蛋白表达的关系. 中华医学杂志, 1998, 78:488-9.
8. 郭飞, 胡伏莲, 贾博琦. 幽门螺杆菌毒素对胃黏膜细胞的c-met、c-myc基因表达的影响. 中华消化杂志,1999;19:137-138.
9. Parsonnet J. Helicobacter pylori and gastric ulcer. Gastroenterol Clin North Am,1993, 22:89-92.
10. Honda S,Fujioka T,Tokieda M,Satoh R, etal. Development of helicobacter pylori –in duodenal ulcer and gastric cancer in Mongolian gerbils. Cancer Res 1998, 58: 4255-4259.
11. Honghton J. Gastric Cancer originating from bone marrow derived cells. Science, 2004, 306: 1568-1571.
12. Mohammed AH, Wilkinson J, Hunt RH. Duodenal ulcer recurrence after helicobacter pylori (H.pylori) eradication: a meta-analysis. Gastroenterology, 1994, 106: A142.
13. 胡伏莲, 黄志烈, 王菊梅, 等. 幽门螺杆菌的根除及其在十二指肠溃疡愈合和复发中的作用. 中华消化杂志, 1996, 16(2): 106-107.
14. Goodwin CS. Duodenal ulcer, Campylobacter pylori, and the “leaking roof” concept. Lancet, 1988, 2: 1467-1469.
15. Levis S, Beardshall K, Haddad G, et al. Campylobacter pylori and duodenal ulcers: the gastrin link. Lancet, 1989, 1: 1167-1168.
16. Peura DA. Ulcerogenesis: intergrating the role of Helicobacter pylori and acid secretion in duodenal ulcer. AM J Gastroenterol. 1997; 92(4 Suppl): 85.
17. 胡伏莲, 贾博琦, 谢鹏雁, 等. 用抗生素治疗合并幽门弯曲菌感染的难治性十二指肠溃疡病. 中华内科杂志, 1988, 27(4): 205-207.
18. Isaacson P, Wright DH. Malignant lymphoma of mucosa-associated lymphoid tissue. A distinctive type of B-cell lymphoma. Cancer, 1983, 52: 1410-1416.
19. Genta RM, Hamner HW, Graham DY. Gastric lymphoid ollicles in Helicobacter pylori infection: frequency, distribution and response to triple therapy. Hum Pathol, 1993, 24: 577-583.
20. Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG.. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet, 1991, 338: 1175-1176.
21. Isaacson PG, Spencer J. Malignant lymphoma of mucosa associated lymphoid tissue. Histopathology, 1987, 11: 445-462.
22. S. Nakamura HKM. Lymphoma of stomach. WHO Classification of Tumors of the Digestive system, 2010, 86
23. Wotherspoon AC, Doglioni C, Diss TC, Pan LX, Moschini A, de Boni M, Isaacson PG.. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue after eradication of Helicobacter pylori, Lancet, 1993, 342: 575-577.
24. Fischbach W, Goebeler-Kolve ME, Dragnosics B, etal. Long term outcome of patients with gastric marginal zone B cell lymphoma of mucosa associated lymphoid tissue (MALT) following exclusive Helicobacter pylori eradication therapy: experience from a large prospective series. Gut, 2004, 53: 34-37.
25. 中华医学会消化病分会幽门螺杆菌学组/幽门螺杆科研协作组.第四次全国幽门螺杆菌感染处理共识报告.中华消化杂志2012,32(10)655-661.
26. Malfertheiner P, Megraud F, O'Morain C, et al. Management of Helicobacter pylori infection--the Maastricht IV/ Florence Consensus Report. Gut, 2012, 61:646-664.