
双逞记-幽门螺杆菌CagA和VacA毒素的致病宿主调制通路
A Tale of Two Toxins: Helicobacter Pylori CagA and VacA Modulate Host Pathways that Impact Disease
Kathleen R. Jones,1 Jeannette M. Whitmire,1 and D. Scott Merrell1,*
幽门螺杆菌感染超过50%的世界人口,是一种导致巨大的医疗负担的致病细菌。幽门螺杆菌感染与多种疾病相关联,例如胃炎、消化性溃疡和胃癌的两种形式:胃腺癌和黏膜相关淋巴组织淋巴瘤。这种相关代表着一种全新的癌症发展;提示幽门螺杆菌是目前唯一的被视为是一种致癌物质的细菌。因此,大量的研究进行了识别细菌因素和负责进展到更严重的疾病状态的异常管制的宿主细胞通路。两个有牵连在这一过程的毒力因子是细胞毒素相关基因 (CagA) 和空泡毒素(VacA),他们分别参与幽门螺杆菌对于细胞的注入和分泌。这两个毒力因素多态性,影响大量的宿主细胞通路。这些复合的因素因为各种 CagA 和 VacA 基因差异而容易造成人群的疾病的严重程度的差异。因此我们着重讨论了这些重要的毒素造成的多种类型的细胞通路和过程。
Front Microbiol. 2010; 1: 115.
Published online 2010 Nov 23. Prepublished online 2010 Aug 25.doi: 10.3389/fmicb.2010.00115
转引自PMCID: PMC3109773
Copyright © 2010 Jones, Whitmire and Merrell.
This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
Helicobacter pylori is a pathogenic bacterium that colonizes more than 50% of the world's population, which leads to a tremendous medical burden. H. pylori infection is associated with such varied diseases as gastritis, peptic ulcers, and two forms of gastric cancer: gastric adenocarcinoma and mucosa-associated lymphoid tissue lymphoma. This association represents a novel paradigm for cancer development; H. pylori is currently the only bacterium to be recognized as a carcinogen. Therefore, a significant amount of research has been conducted to identify the bacterial factors and the deregulated host cell pathways that are responsible for the progression to more severe disease states. Two of the virulence factors that have been implicated in this process are cytotoxin-associated gene A (CagA) and vacuolating cytotoxin A (VacA), which are cytotoxins that are injected and secreted by H. pylori, respectively. Both of these virulence factors are polymorphic and affect a multitude of host cellular pathways. These combined facts could easily contribute to differences in disease severity across the population as various CagA and VacA alleles differentially target some pathways. Herein we highlight the diverse types of cellular pathways and processes targeted by these important toxins.
Keywords: Helicobacter, CagA, VacA, cell-signaling, toxin, gastric cancer
Introduction
Helicobacter pylori is a rather recently discovered bacterium that was first isolated from a gastritis patient in 1982 (Marshall and Warren, 1984). H. pylori is a Gram-negative, spiral shaped, and fastidious microaerophile that inhabits the inhospitable environment of the human stomach. In this niche, it can persist, if left untreated, for the lifetime of its host (Blaser, 1990). The infection rates for H. pylori vary widely geographically; however, overall this bacterium colonizes the gastric mucosa of over 50% of the world's population (The EUROGAST Study Group, 1993; Matysiak-Budnik and Megraud, 1997).
The majority of people colonized with H. pylori develop subclinical gastritis. However, a small percentage of colonized individuals progress to clinical disease, the majority of which will present with gastritis. In some individuals, colonization results in more severe disease manifestations. H. pylori is the etiological agent of peptic ulcer disease, 75% of gastric ulcers and 90% of duodenal ulcers are attributed to H. pylori infection (Ernst and Gold, 2000), as well as two distinct forms of gastric cancer: mucosa-associated lymphoid tissue (MALT) lymphoma and adenocarcinoma (Parsonnet et al., 1991, 1994; Talley et al., 1991; Blaser, 1998). Due to this correlation, H. pylori is the only bacterium currently classified as a class I carcinogen by the World Health Organization (“Infection with Helicobacter pylori,” 1994). Indeed, gastric cancer is still the second most common cause of worldwide cancer mortality, and this high rate could be reflective of the high incidence of H. pylori infection (Neugut et al., 1996; Yamamoto, 2001; Parkin et al., 2005; Crew and Neugut, 2006).
Due to H. pylori's association with a variety of severe gastric diseases, many studies have been conducted to elucidate the bacterial, host, and environmental factors that impact disease progression. To date, several bacterial virulence factors have been associated with gastric cancer. For instance, the outer membrane proteins, HomB (Jung et al., 2009; Oleastro et al., 2009) and BabA – in Western type cytotoxin-associated gene A (CagA) containing strains (Gerhard et al., 1999), but not in East Asian type CagA containing strains (Mizushima et al., 2001) have been associated with progression to gastric cancer, while OipA (Odenbreit et al., 2009; Ben Mansour et al., 2010) and DupA (Lu et al., 2005; Douraghi et al., 2008; Nguyen et al., 2009; Schmidt et al., 2009; Hussein, 2010) have more controversial roles in disease development. The effectors, CagA and vacuolating cytotoxin A (VacA), have also been shown to influence disease state, and are probably the most well studied virulence factors of H. pylori. These toxins have been shown to have multiple effects on host cells, as well as to modulate multiple cellular pathways in what appears to be a complex orchestration that ultimately leads to disease. To begin to shed some light on these pathways, as well as on the etiology of disease, this review will highlight some major findings regarding CagA, VacA, and their specific effects on host cells. Due to the large amount of literature on this subject and space limitations, an exhaustive review is not provided. However, we encourage readers to explore the excellent reviews by Cover and Blanke (2005), Hatakeyama and Higashi (2005), and Rieder et al. (2005a).
Cytotoxin-Associated Gene A
CagA is arguably the most well studied virulence factor of H. pylori. It is encoded on the cag pathogenicity island, which is a horizontally acquired 40 kb DNA segment that encodes for a type IV secretion system, and is the only known effector protein to be injected into host cells (Censini et al., 1996; Akopyants et al., 1998).cagA is the last gene on the cag pathogenicity island, and encodes for the 120–145 kDa immunodominant CagA protein (Covacci et al., 1993; Tummuru et al., 1993). Since its discovery, CagA has been shown to impact disease, especially more severe disease states like gastric cancer (Blaser et al., 1995; Kuipers et al.,1995; Xiang et al., 1995; Parsonnet et al., 1997; Huang et al., 2003). cagA is present in ∼70% of strains worldwide, but this rate varies geographically from between 90–95% in East Asian countries (South Korea, China, Japan) and only about 40% in Western countries (Australia, United States of America, England; Hatakeyama, 2006).
Once injected into host cells, CagA can act directly in an unphosphorylated state to influence cellular tight junction (Amieva et al., 2003; Bagnoli et al., 2005; Murata-Kamiya et al., 2007; Oliveira et al., 2009), cellular polarity (Saadat et al., 2007; Zeaiter et al., 2008), cell proliferation and differentiation (Mimuro et al.,2002; Chang et al., 2006; Murata-Kamiya et al., 2007; Lee et al., 2010), cell scattering (Mimuro et al., 2002), induction of the inflammatory response (Brandt et al., 2005), and perhaps cellular elongation (Figure (Figure1;1; Saadat et al., 2007; Umeda et al., 2009). Moreover, upon entering the eukaryotic cell, CagA localizes to the plasma membrane where it can be phosphorylated by either Abl kinase or Src family kinases (Stein et al.,2000, 2002; Selbach et al., 2002; Poppe et al., 2007; Tammer et al., 2007). These kinases phosphorylate tyrosine residues found in a five amino acid repeat, Glu-Pro-Ile-Tyr-Ala (EPIYA), within the carboxy-terminus of CagA (Higashi et al., 2002a,b). These repeats can be categorized based on the amino acid sequences found within the regions flanking the EPIYA sequence to yield four distinct EPIYA motifs, which are known as EPIYA-A, -B, -C, and -D. Two combinations of these motifs predominate: Western CagA, which contains EPIYA-A, -B, and -C motifs (strains have been genotyped that contain up to five -C motifs) and East Asian CagA, which contains EPIYA-A, -B, and -D motifs (Figure (Figure1;1; Covacci et al., 1993; Higashi et al., 2002a,b; Stein et al., 2002; Naito et al., 2006; Argent et al., 2008a; Nguyen et al., 2008). Additionally, there is a multimerization motif that consists of a 16 amino acid sequence present within the EPIYA repeat region (Ren et al., 2006). Once phosphorylated, CagA can form a complex with the CT10 regulator of kinase (Crk) adaptor protein (Suzuki et al., 2005; Brandt et al., 2007), Abl kinase and a splice variant of Crk, CrkII (Tammer et al., 2007), or the Src homology 2 phosphatase (SHP-2; Higashi et al., 2002b). Each of these interactions influences cellular shape and motility (Higashi et al., 2002b; Suzuki et al., 2005; Brandt et al., 2007; Tammer et al., 2007). CagA that is phosphorylated at the primary phosphorylation sites (EPIYA-C and -D) shows varying affinities for SHP-2 based on the particular EPIYA variant as well as subsequent differential effects on the pathways influenced by the phosphorylated CagA/SHP-2 complex (Higashi et al.,2002a,b).
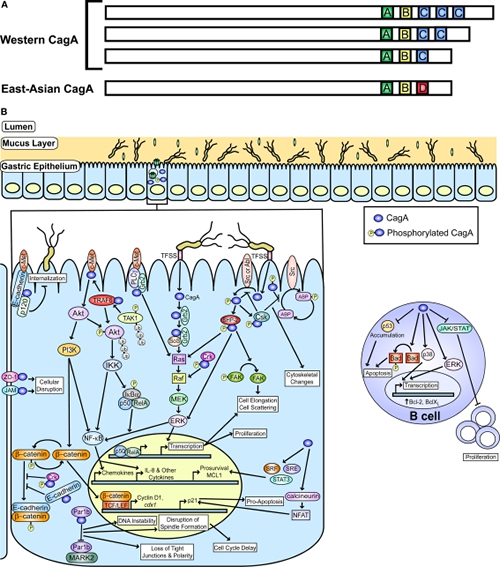
Figure 1
CagA and known host cell targets. (A) A schematic representation of CagA with the polymorphic region containing different EPIYA motif (A, B, C, and D) combinations is shown and is adapted from Hatakeyama and Higashi (2005). (B) A graphic depiction of the gastric mucosa and known host pathways impacted by phosphorylated and non-phosphorylated CagA is shown. Pathways targeted in epithelial cells and B cells are indicated. The actin binding proteins (ABP) affected by CagA include vinculin, cortactin, and ezrin. This figure was adapted from an earlier version by Rieder et al. (2005a).
CagA and disease
The mere presence of CagA is associated with more severe disease forms (Crabtree et al., 1991; Covacci et al., 1993; Blaser et al., 1995; Gwack et al., 2006). In fact, cancer patients are at least twice as likely to be infected with an H. pylori strain that is cagA positive than one that is cagA negative (Blaser et al., 1995; Gwack et al., 2006). It has additionally been demonstrated in vivo that cagA plays an important role in disease progression in a Mongolian gerbil model where gastric cancer develops within 12 weeks (Watanabe et al., 1998; Wirth et al., 1998; Ogura et al., 2000; Peek Jr. et al., 2000; Franco et al., 2005, 2008; Rieder et al., 2005b). The differences in affinity of various EPIYA motifs for SHP-2 and subsequent differences in induction levels of downstream pathways has been speculated to impact the differences in disease rates, especially gastric cancer (Higashi et al., 2002a,b; Hatakeyama, 2004). In fact, increasing numbers of EPIYA-C motifs have been suggested to be associated with cancer development (Azuma et al., 2002; Satomi et al.,2006), and epidemiological studies have identified a correlation between increasing number of -C motifs and heightened disease severity (Yamaoka et al., 1999; Higashi et al., 2002a; Argent et al., 2004). Since EPIYA-D has the strongest affinity for SHP-2 (Higashi et al., 2002a), it is not surprising that East Asian CagA produces more inflammation and atrophy (Azuma et al., 2004) as well as greater morphological changes in infected cells (Higashi et al., 2002a). Moreover, the variability in CagA is important when analyzing the geographic areas with the highest gastric cancer rates; these areas not only have the highest colonization rates, but also contain the highest percentage of H. pylori strains that carry the cagA gene, in particular the EPIYA-ABD allele (Ahn et al., 1991; Yamamoto, 2001; Crew and Neugut, 2006; Hatakeyama, 2006). Indeed, we identified an association between gastric cancer development and EPIYA-ABD CagA through a large scale molecular epidemiological study of strains from South Korea (Jones et al., 2009).
CagA Phosphorylation Independent Events
Physical effects on host cells
Despite the importance of CagA phosphorylation, CagA has many effects on the host cell, and some of these effects are accomplished in a phosphorylation independent manner. One of the most noticeable CagA dependent effects on host cells is the disruption of tight junctions and induction of changes in cell morphology. CagA has been shown to affect cellular tight junctions in a phosphorylation independent manner (Amieva et al., 2003; Bagnoli et al., 2005; Murata-Kamiya et al., 2007; Oliveira et al., 2009), and has been shown to be important for the recruitment of the junctional adhesion molecule (JAM) and the tight junction protein, zona occludens-1 (ZO-1) to points of bacterial contact (Amieva et al., 2003; Bagnoli et al.,2005). Murata-Kamiya et al. (2007) showed by immunoprecipitation that E-cadherin physically interacts with both wild type and phosphorylation resistant variants of CagA, and that this interaction inhibits the association of E-cadherin with β-catenin, which subsequently results in the accumulation of nuclear and cytoplasmic β-catenin.
Additionally, it has been demonstrated that CagA binds to and prevents the kinase activity of the partitioning-defective 1/microtubule affinity-regulating kinase (Par1b/MARK2), thereby escalating the loss of tight junctions and polarity (Saadat et al., 2007; Zeaiter et al., 2008). CagA binds to Par1b, as well as other members of this kinase family, through the multimerization sequence (Saadat et al., 2007; Lu et al., 2008), specifically 14 of the 16 amino acids of the multimerization motif are required (FPLKRHDKVDDLSK; Nesic et al., 2010). This interaction has been suggested to contribute to host cell elongation (Saadat et al.,2007; Umeda et al., 2009). East Asian CagA binds Par1b with a stronger affinity than CagA from Western strains, and the efficiency and strength of binding to Par1b among Western strains appears proportional to the number of Western multimerization sequences (Lu et al., 2008). In addition to affects on cell elongation and disruption of cellular junctions, interaction of CagA and Par1b also causes spindle dysfunction, which delays progression from prophase to metaphase and is hypothesized to result in DNA instability (Lu et al., 2009).
Moreover, recent evidence has shown that CagA has an effect on how invasive H. pylori can be. Though considered an extracellular pathogen, numerous studies have shown that H. pylori is able to invade and survive inside host cells (Amieva et al., 2002; Semino-Mora et al., 2003; Oh et al., 2005). Upon infection with cagA positive H. pylori strains, a multiprotein complex is formed through the association of CagA, c-Met, E-cadherin, and p120-catenin, and this complex influences whether the bacteria can become intracellular (Oliveira et al., 2009). When this complex is present in a cell line that H. pylori can normally invade, it suppresses the ability of H. pylori to be internalized (Oliveira et al., 2009).
Cellular differentiation
Since there is a causal link between gastric cancer, H. pylori infection, and the presence of cagA, there is no doubt that effects on host cell differentiation and proliferation are important for ultimate disease progression. In keeping with this, cagA positive strains of H. pylori influence a factor with known oncogenic potential, β-catenin (El-Etr et al., 2004; Franco et al., 2005). β-catenin has two distinct functions, it links cadherins with the actin cytoskeleton and is part of the WNT signaling pathway (Tolwinski and Wieschaus, 2004). When unphosphorylated CagA binds E-cadherin, it prevents the formation of the E-cadherin/β-catenin complex, which ultimately leads to accumulation of β-catenin in both the nucleus and cytoplasm (Murata-Kamiya et al., 2007). Kurashima et al. (2008) showed that while phosphorylation of the EPIYA motifs was not necessary for deregulation of β-catenin, the CagA multimerization sequence was necessary. However, this process is likely multifactorial and complex since some evidence indicates that E-cadherin dissociation is independent of CagA and that the E-cadherin/β-catenin/p120ctn complex is not affected to the same degree in all studies (Weydig et al., 2007). When the E-cadherin/β-catenin complex is disrupted, cytoplasmic β-catenin is dephosphorylated and then translocates to the nucleus, where it forms heteromers with other transcription factors and transcribes a number of genes with oncogenic potential (Lickert et al., 2000; Murata-Kamiya et al., 2007). In fact CagA was found to upregulate the β-catenin-dependent cdx1 gene (Lickert et al., 2000; Murata-Kamiya et al., 2007), which encodes a transcription factor important for transdifferentiation of intestinal cells (Mizoshita et al., 2004), as well as to affect the expression of goblet-cell mucin MUC2, an intestinal-differentiation marker (Murata-Kamiya et al., 2007); both are indicators of gastric intestinal metaplasia.
Cell cycle, survival, and proliferation
Increased cellular proliferation is one indicator of cancer that has been demonstrated to result from infection with cagA positive strains of H. pylori (Peek Jr. et al., 1997, 1999). This increased proliferation can happen through CagA mediated activation of the ERK/MAPK pathway. CagA activates ERK through interaction with growth factor receptor bound 2 (Grb2), and appears to interact with both phosphorylated and non-phosphorylated CagA (Mimuro et al., 2002; Selbach et al., 2009). However, it should be noted that the phosphorylation sequences themselves are essential for Grb2 binding to non-phosphorylated CagA (Mimuro et al., 2002), which similar to the strategy for binding of CagA to SHP-2 (Li et al., 1993), likely occurs through CagA binding to the Grb2 SH2 domains. In a normal cell, upon receiving an extracellular signal, Grb2 binds son of sevenless (SoS), which increases the formation of the Ras-GTP complex and activates the Raf → MEK → ERK pathway. This pathway increases activation of transcription factors involved in cell proliferation (Gale et al., 1993). In CagA intoxicated cells, CagA can bind Grb2, and then the CagA/Grb2/SoS complex activates the Ras → ERK pathway as described above (Mimuro et al., 2002).
Activity of the serum response factor and serum response element (SRF and SRE, respectively) transcription factors are also increased in CagA transfected cells in a CagA phosphorylation independent manner (Hirata et al., 2002). Activation of SRE appears to be mediated by increased DNA-binding by the E-26 like protein-1 (Elk1; Hirata et al., 2002). Evidence of the role of CagA in SRE/SRF activation can be found through CagA mediated increases in levels of the anti-apoptotic protein myeloid cell leukemia sequence-1 (MCL1), which acts as a pro-survival factor (Mimuro et al., 2007). Furthermore, the tendency of CagA to affect transcription factor activity seems to be a common theme since the signal transducer and activator of transcription 3 (STAT3) pathway, which induces cellular proliferation (Klampfer, 2006), has been shown to be induced in vitro and in vivo in a CagA dependent but phosphorylation independent manner (Bronte-Tinkew et al., 2009). Moreover, it was recently confirmed that non-phosphorylated CagA preferentially activates STAT3 (Lee et al., 2010). The activation of so many transcription factors by CagA is evidence of the broad influence of CagA on a wide variety of cellular functions.
CagA appears to influence the development of MALT lymphoma in a phosphorylation independent manner; CagA can inhibit apoptosis of B cells through inhibition of the accumulation of p53 due to decreased p53transcription (Umehara et al., 2003). Interestingly, B cell survival is also likely due in part to an increase in phosphorylated ERK1/2 (Zhu et al., 2007), which when moderately increased can inhibit apoptosis and promote proliferation (Park et al., 2004). However, it should be noted that transfection with CagA also leads to phosphorylation of the pro-apoptotic protein, Bad (Zhu et al., 2007). Conversely, translocation of CagA by H. pylori results in upregulation of both the ERK and p38 pathways, which lead to upregulation of the pro-survival proteins, Bcl-2 and Bcl-XL (Lin et al., 2010). Clearly, CagA has an effect on cell survival, and most of the literature suggests that CagA can inhibit apoptosis of B cells, which likely promotes the development of MALT lymphoma.
Conversely, ectopically expressed CagA can also suppress cellular proliferation in IL-3-dependent B-lymphoid cells through suppression of JAK-STAT signaling (Umehara et al., 2003). Moreover, transfection of AGS cells with CagA results in increased expression of the pro-apoptotic factor p21WAF1/Cip1 due to CagA mediated nuclear translocation of the nuclear factor of activated T cells family transcription factor (NFATc3; Yokoyama et al., 2005). Interestingly, p21WAF1 expression can also occur as a direct result of excessive ERK1/2 activation (Bhat and Zhang, 1999; Park et al., 2005; Yun et al., 2005). Finally, CagA induced deregulation of β-catenin, increases the expression of Cyclin D1 (Udhayakumar et al., 2007), which influences progression of cells from G1 to S phase; thereby promoting cell survival in a CagA phosphorylation-independent manner (Chang et al., 2006; Murata-Kamiya et al., 2007). CagA obviously affects cell survival, proliferation, and differentiation, all of which can affect the progression of disease, including development of gastric cancer.
Cell scattering
In order for cancer cells to spread or metastasize, they must detach and scatter to a new area. CagA increases cell scattering by targeting the hepatocyte growth factor receptor (c-Met), which acts as an adaptor molecule for proteins like Grb2, phospholipase Cγ (PLCγ), and STAT3 (Mimuro et al., 2002; Churin et al., 2003). CagA/Grb2/SoS → Ras-GTP complex → Raf → MEK → ERK signaling leads not only to an increase in transcription factors that promote cellular proliferation, but also to an increase in cell scattering (Mimuro et al., 2002). In support of this, cell scattering due to H. pylori infection is suppressed by blocking PLCγ activity (Churin et al., 2003). Though once again, the role of CagA in this process is complex; some work suggests no association between enzymatic activity of PLCγ and the cagA status of H. pylori strains (Bode et al., 2001). However, it is clear that non-phosphorylated CagA interacts with c-Met through interaction with the multimerization domain (Suzuki et al., 2009). The consequence of this interaction is activation of phosphatidylinositol 3-kinase (PI3K) signaling through Akt, which subsequently activates NF-κB and β-catenin (Suzuki et al., 2009). It has been suggested that CagA binds to c-Met and then recruits TNF receptor associated factor 6 (TRAF6; Sun et al., 2004) and poly-ubiquitinated transforming growth factor-β-activating kinase 1 (TAK1; Lamb et al., 2009; Lamb and Chen, 2010). TRAF6 then activates Akt (Yang et al., 2009), which potentially activates NF-κB and RelA through activation of the IκB kinase (IKK) complex (Suzuki et al., 2009; Takeshima et al., 2009). CagA modulates multiple pathways that impact cell scattering, and these pathways that are activated appear to have multiple downstream targets that can affect numerous cellular processes in addition to cell scattering.
Inflammation
A hallmark of H. pylori infection is increased and chronic inflammation. This appears to occur due to activation of NF-κB and persistent induction of IL-8. While it remains controversial (Crabtree et al., 1995; Lamb et al., 2009; Schweitzer et al., 2010), this IL-8 induction has been shown to be CagA dependent through studies that ectopically expressed CagA or various CagA EPIYA motifs (Brandt et al., 2005; Kim et al., 2006), through IL-8 promoter reporter assays (Sharma et al., 1998), and through analysis of inflammation and NF-κB activation in Mongolian gerbils infected with cagA positive and cagA negative H. pylori strains (Shibata et al., 2006). Moreover, Keates et al. (1999) showed that IL-8 secretion is affected by the activation of MAPKs by cagA positive H. pylori strains. It has been demonstrated that activation of NF-κB and induction of IL-8 occurs through the activation of the Ras → Raf → Mek → ERK → NF-κB pathway and is independent of SHP-2 or c-Met (Brandt et al., 2005). Indeed, Brandt et al. (2005) demonstrated that IL-8 induction was CagA phosphorylation independent. Kim et al. (2006) confirmed that NF-κB activation and subsequent induction of IL-8 were due to activation of the MAPK pathways and also analyzed the role of the different EPIYA motifs by analysis of transfected cells with CagA constructs that differed only in the EPIYA region. Analysis of Western CagA-specific sequences and East Asian CagA-specific sequences revealed that the levels of IL-8 induction are not significantly different between the CagA variants. However, it should be noted that Argent et al. (2008a) demonstrated that CagA related differences in IL-8 induction were dependent on the EPIYA motifs and that strains containing East Asian CagA induce the greatest levels of IL-8. However, they did not investigate the influence of phosphorylation status on these differences. Since persistent inflammation is a hallmark of H. pylori infection and is linked to more severe diseases, and since CagA affects the inflammatory process, it is easy to understand what an important role CagA plays in persistent infection and disease development.
CagA Phosphorylation Dependent Events
Targeting of SHP-2
The most striking H. pylori induced morphological change to host cells is the induction of the “hummingbird phenotype,” which occurs as a direct result of phosphorylated CagA complexing with SHP-2 and subsequent increased ERK1/2 activation. Normally, SHP-2 functions to increase cellular proliferation and motility and is activated by interacting with a phosphorylated Gab protein (Neel et al., 2003). CagA has been shown to be able to mimic the function of the eukaryotic Gab protein (Hatakeyama, 2003; Botham et al., 2008). Both in vitro and in vivo (Yamazaki et al., 2003), CagA forms a complex with SHP-2 after phosphorylation of an EPIYA-C or -D motif (Higashi et al., 2002a). The formation of this complex, as well as the subsequent deregulation of SHP-2 as a means of CagA mediated effects on gastric cancer, is of relevance since mutations within the gene encoding for SHP-2 (PTPN11) have been identified in multiple forms of cancer (Tartaglia et al., 2003; Bentires-Alj et al., 2004). Additionally, there is an increase in the risk of gastric cancer development in H. pylori infected patients with certain PTPN11 polymorphisms (Goto et al., 2006). Again, this demonstrates the potential of CagA to impact disease progression.
Activation of ERK
The ERK MAP kinases are activated in a CagA phosphorylation dependent manner upon infection with H. pylori cagA positive strains, leading to a SHP-2 dependent change in cell motility and morphology (Neel et al., 2003). Indeed, inhibition of the phosphorylation of CagA, knockdown of SHP-2 expression, or the disruption of the CagA/SHP-2 complex abrogates cell elongation; thus, indicating that the “hummingbird phenotype” is a product of the SHP-2/CagA complex (Segal et al., 1999; Higashi et al., 2002b, 2004; Higuchi et al., 2004). In fact, this complex activates the ERK pathway by activating Rap1 → B-Raf → ERK and has been proven to activate ERK in both a Ras independent and dependent manner (Higashi et al.,2004). In addition, CagA promotes cell proliferation through activation of ERK, which subsequently promotes progression through the cell cycle (Roovers and Assoian, 2000; Tsutsumi et al., 2003).
Recent data indicates that the phosphorylation status of CagA may act as a signaling switch between the JAK/STAT3 and SHP-2/ERK pathways. This process is mediated through gp130 (Lee et al., 2010). Unphosphorylated Cag activates STAT3, while phosphorylated CagA preferentially activates ERK1/2 phosphorylation (Lee et al., 2010). This differential activation based on phosphorylation status illustrates the complexity of the effects that CagA has on the host cell.
Non-ERK mediated cytoskeletal rearrangement and scattering
Important steps in the creation of elongated cells include the decrease in cellular adhesions and the deregulation of the actin-binding proteins that maintain proper cellular shape (Moese et al., 2004; Bourzac et al., 2007). Phosphorylated CagA binding and activation of SHP-2 leads to increased tyrosine dephosphorylation of the focal adhesion kinase (FAK; Tsutsumi et al., 2006), which is important for cellular elongation of host cells; when dominant-negative FAK is expressed, host cells change morphology, while constitutively active FAK abrogates the formation of this morphological change (Tsutsumi et al., 2006). Additionally, studies have identified multiple actin-binding proteins that, when tyrosine-dephosphorylated, promote CagA phosphorylation dependent cell elongation. These include vinculin (Moese et al., 2007), cortactin (Selbach et al., 2003; Selbach and Backert, 2005), and ezrin (Selbach et al., 2004). Since the SHP-2 phosphatase is not required for the dephosphorylation of cortactin, the dephosphorylation of these actin-binding proteins is likely a result of blocked activity of a kinase, and therefore a product of the phosphorylated CagA negative feedback loop that inhibits the Src kinase (Selbach et al., 2003). In fact, this is the case for ezrin as inhibition of Src family kinases increases dephosphorylation of ezrin (Selbach et al.,2004), and host elongation is achieved simply through inactivation of Src, which results in dephosphorylation of all Src substrates, including vinculin, ezrin, and cortactin (Backert et al., 2010). This inhibition of Src could occur either directly or through the recruitment of C-terminal Src kinase (Csk), as described in the next section (Selbach et al., 2003; Tsutsumi et al., 2003; Backert et al., 2010). Finally, phosphorylated CagA can bind Crk adaptor proteins (Crk-I, Crk-II, and Crk-L; Suzuki et al., 2005). This interaction is important for cell scattering, disruption of E-cadherin/catenin, and activation of Raf (Suzuki et al., 2005). Furthermore, it was recently shown that CagA phosphorylation could occur via Abl instead of Src, thereby activating downstream effects, specifically cell scattering and motility (Poppe et al., 2007; Tammer et al., 2007). Moreover, Abl could also form a complex with CrkII and CagA (Tammer et al., 2007). Taken together, these findings show that phosphorylation of CagA is very important for host cell shape and adhesion. This fact implicates the degree of phosphorylation as a consequence of the cagA allele carried by a strain as being important for development of gastric carcinomas.
CagA feedback loop, Src vs. Csk
As mentioned above, CagA participates in a negative feedback loop that allows for regulation of the amount of phosphorylated CagA. CagA can bind Csk via direct interaction with the EPIYA-A and -B motifs (Tsutsumi et al., 2003, 2006). Formation of this complex leads to the inhibition of the Src family kinases (SFKs) through Csk tyrosine phosphorylation of an inhibitory C-terminal residue on Src (Tsutsumi et al.,2003, 2006; Hatakeyama, 2004). CagA can also directly inhibit SFK activity (Selbach et al., 2003). While the purpose of this negative feedback system is not completely clear, it appears that in its absence, CagA is excessively toxic to cells (Tsutsumi et al., 2003; Hatakeyama, 2004, 2006). Thus, this loop has been hypothesized to promote long-term colonization of cagA positive H. pylori strains (Hatakeyama, 2006).
Interactions with unknown function
Recent proteomic screens identified a number of proteins that appear to interact with phosphorylated CagA (Selbach et al., 2009). These include PI3K, Grb2, Ras-GAP, Grb7, and Shp1. The consequences of these interactions with phosphorylated CagA is still unknown (Selbach et al., 2009). However, it is clear that both Grb2 and PI3K are actively involved in H. pylori pathogenesis when CagA is not phosphorylated (Mimuro et al., 2002; Suzuki et al., 2009). Indeed, unphosphorylated CagA is known to bind to Grb2, which activates ERK signaling, and leads to increased cellular proliferation, transcription factor activation, cell scattering (Mimuro et al., 2002), and activation of Akt and PI3K signaling. These activation events subsequently stimulate both the β-catenin and NF-κB pathways (Suzuki et al., 2009). So the interaction of some of these proteins with phosphorylated CagA may represent redundant mechanisms of action.
CagA Independent/Redundancy
Inflammation appears important for H. pylori growth in vivo. For instance, H. pylori induced inflammation results in a decrease in the inhibitor of gastrin; gastrin has been proven to be a H. pylori growth factor (Levi et al., 1989; Moss et al., 1992; Chowers et al., 2002; Blaser and Atherton, 2004). Given the importance of inflammation for H. pylori colonization and persistence, there are redundant mechanisms by which H. pyloriinduces inflammation. For instance, in addition to CagA effects, NF-κB activation can also be achieved through TLR4 recognition of LPS (Ohmae et al., 2005; Lamb and Chen, 2010) or through type IV secretion system (T4SS) delivered peptidoglycan (PG) binding to nucleotide-binding oligomerization domain 1 (NOD1; Figure Figure2;2; Viala et al., 2004; Kaparakis et al., 2010). The inflammatory response caused by the interaction of peptidoglycan-NOD1 may be a result of the activation of AP-1 (Allison et al., 2009), which functions as a transcription factor for cytokines and chemokines such as IL-8 (Faisst and Meyer, 1992; Jain et al., 1992; Chauhan et al., 1994; Rao, 1994). Recently, it has also been demonstrated that the binding of NOD1 to its ligand, in this case PG, activates RICK, which allows RICK to interact directly with TRAF3 followed by TRAF3 interaction with TBK1 (Watanabe et al., 2010). TBK1, as well as IKKε, leads to the production of cytokines, such as the type 1 interferons (IFN) like IFN-β. The production of IFN-β is responsible for NOD1’s ability to increase the level of the chemokine IP-10 as well as the induction of and nuclear translocation of the transcription factor interferon-stimulated gene factor 3 (ISGF3; Watanabe et al.,2010). The presence of the cag pathogenicity island has also been shown to lead to increased inflammation (Censini et al., 1996); 14 of the 27 genes within the island are essential for IL-8 induction (Fischer et al.,2001). With multiple bacterial factors that induce NF-κB, this begs the question of why the bacterium needs mechanisms for such redundancy. Lamb and Chen (2010) postulate that since CagA and PG target different cellular signaling molecules, they may synergistically activate NF-κB, and that this synergy may be important. Alternately, in strains where CagA is not present or is not a potent inducer of IL-8 and NF-κB, PG may serve as the major inducer of the inflammatory response. Both of these hypotheses highlight the importance of the induction of an inflammatory response for H. pylori, probably due to the requirement for gastrin or other nutrients.
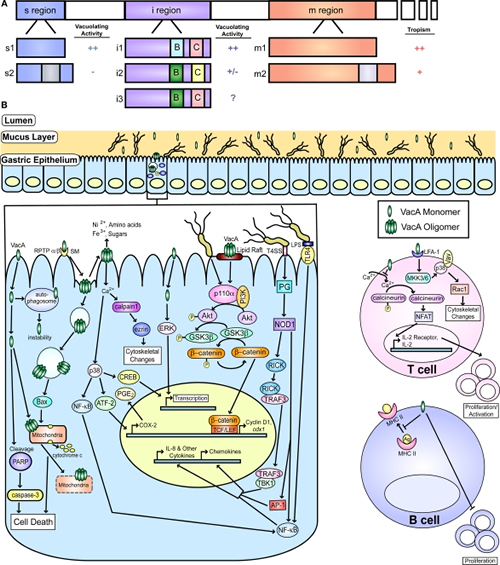
Figure 2
VacA and known host cell targets. (A) A schematic representation of VacA with the three major regions of polymorphisms (s, i, and m) is shown. Additionally, schematics of the known alleles of each region are shown. The i region contains two important polymorphic regions known as Cluster B and Cluster C, which are designated by a B and C, respectively on the diagram. The activity attributed to each of the regions of the toxin (vacuolating activity or cellular tropism) are indicated, and the impact of each allele on these effects is shown. The highest level of activity or the broadest tropism is defined as ++, intermediate tropism is indicated by a +, low activity is indicated as a +/−, no activity is designated by a −, and incomplete information is indicated by a ?. (B) A depiction of the gastric mucosa and known host pathways targeted by VacA is shown. One of the receptors, sphingomyelin is designated by SM. Pathways targeted in epithelial cells and B and T cells are indicated. Additionally, activation of several pathways by peptidoglycan (PG) and LPS are shown. This figure was adapted from an earlier version by Rieder et al. (2005a).
Vacuolating Cytotoxin A
VacA is another important factor that has been indicated to have effects on H. pylori virulence and to target numerous host cell pathways (Figure (Figure2).2). Activity of this protein was found when H. pylori filtrates were shown to induce large host cell vacuoles (Leunk et al., 1988). The VacA cytotoxin appears to be produced and secreted by most, if not all, H. pylori strains, but possesses no similarity to any other known bacterial or eukaryotic protein (Atherton et al., 1999; Cover and Blanke, 2005). Once produced, VacA can remain on the bacterial surface (Ilver et al., 2004) or be secreted as an approximately 88 kDa toxin (Cover and Blaser,1992). Secreted VacA monomers oligomerize (Lupetti et al., 1996; Cover et al., 1997;
Lanzavecchia et al.,1998; Adrian et al., 2002) but dissociate upon exposure to a non-neutral environment. In fact, exposure to alkaline or acidic conditions actually amplifies the activity of VacA (de Bernard et al., 1995; Cover et al.,1997; Molinari et al., 1998a; Yahiro et al., 1999). Once secreted, VacA undergoes proteolytic cleavage to yield two smaller products, p33 and p55. However, to date the consequence of this cleavage is not understood (Telford et al., 1994; Nguyen et al., 2001; Willhite et al., 2002; Ye and Blanke, 2002; Torres et al., 2004). The smaller p33 product and about 100 amino acids of p55 are responsible for the vacuolating activity of VacA (de Bernard et al., 1997, 1998; Ye et al., 1999). The p33 domain is strongly hydrophobic and contains characteristic transmembrane dimerization motifs that are responsible for insertion into the host cellular membrane and vacuolating activity (Vinion-Dubiel et al., 1999; Ye and Blanke, 2000, 2002; McClain et al., 2001a, 2003; Kim et al., 2004), whereas the p55 domain has a crucial role in binding to host cells (Garner and Cover, 1996; Pagliaccia et al., 1998; Reyrat et al., 1999; Wang and Wang, 2000; Wang et al., 2001).
Like CagA, VacA is polymorphic. However, unlike CagA, this variation begins within the amino-terminus of VacA. Three regions of variation have been defined and there are at least two primary variants in each region; the regions are designated as the signal (s), intermediate (i), and middle (m) regions (Figure (Figure2;2; Atherton et al., 1995; Rhead et al., 2007; Chung et al., 2010). The s region of VacA is found in the p33 portion of the toxin and influences vacuolating activity and efficiency of anion channel formation due to the hydrophobic nature of the amino acid residues found near the proteolytic cleavage site (Pugsley, 1993; McClain et al., 2001b). The s2 variant undergoes cleavage at an alternate site, thereby providing an extension of 12 hydrophilic amino acids (Atherton et al., 1995). The s1 variant contains more hydrophobic amino acids near the cleavage site than the s2 variant; thus, the s1 sequence is more easily inserted into the host cell membrane (Letley and Atherton, 2000; McClain et al., 2001b). The m region is found in the p55 portion of the toxin and influences host cell tropism; the m1 region is toxic to a wider range of host cells (Pagliaccia et al., 1998; Ji et al., 2000; Amieva and El-Omar, 2008). The i region is located between the s and m regions and is the most recent region to be described. The i region has been suggested to be the best indicator of disease severity (Rhead et al., 2007) and three primary variants have been identified (Chung et al., 2010). The i1 region is believed to be associated with stronger vacuolating activity and more severe disease states than the i2 region (Rhead et al., 2007). Furthermore, strains carrying VacA s1, i1, m1, and combinations of these alleles are overall associated with more severe disease (Letley et al., 2003; Rhead et al., 2007; Basso et al., 2008; Jang et al., 2010). This association could be due to increased anion channel formation, vacuolating activity, and cell tropism from having the s1, i1, and m1 regions, respectively.
In recent years, a number of studies have elucidated multiple receptors for VacA and shown that VacA uses different receptors based on different host cell types (Sewald et al., 2008a). On epithelial cells, several different receptors have been identified. Among these are RPTPα, which is a receptor-like protein tyrosine phosphatase that appears to be used by VacA on G401 cells (a human kidney tumor cell line) and in AGS cells (an adenocarcinoma cell line; Yahiro et al., 2003; Tegtmeyer et al., 2009). Another receptor which needs to be glycosylated for VacA to bind to it is RPTPβ, which can be used by VacA on AZ-521 (gastric epithelial-derived cells; Yahiro et al., 1999, 2004). When RPTPβ is artificially increased in some cell lines, toxicity to VacA also increases (Padilla et al., 2000). The importance of this receptor in vivo has been demonstrated in RPTPβ knock out mice, which becomes resistant to VacA mediated ulceration (Fujikawa et al., 2003). Additionally, sphingomyelin was recently identified as a receptor that is important for VacA binding and vacuolating activity of the toxin (Gupta et al., 2008, 2010). Finally, VacA can also bind to T cells using the lymphocyte function-associated antigen-1 (LFA-1; Sewald et al., 2008b). The fact that VacA can use different receptors based on the cell type targeted may help explain this toxin's diverse functions. While much is known about the various functions of the toxin, relatively little is currently known about the exact host signaling pathways affected by the toxin. Thus, herein we discuss what is currently known about the major cellular processes affected by VacA.
VacA Functions
Anion channel formation and vacuolation
VacA can oligomerize within the plasma membrane and can cause formation of anion-selective channels (Szabo et al., 1999). These channels may be used to increase the efflux of complex molecules, such as bicarbonate and urea, out of the host cell (Szabo et al., 1999; Debellis et al., 2001; Tombola et al., 2001), which may aid H. pylori growth (Montecucco and Rappuoli, 2001). In addition to forming anion-selective channels in vitro, VacA can reduce the transepithelial electric resistance of polarized cells by increasing paracellular epithelial permeability. This allows the release of some cations, such as Fe3+ and Ni2+, as well as more complex molecules such as amino acids and sugars (Papini et al., 1998).
One of the most striking effects of VacA on host cells is the creation of large cytoplasmic vacuoles that contain the markers for late endosomes and lysosomes (Molinari et al., 1997). Once VacA is internalized by the host cell, it is trafficked to the early endosome by F-actin containing structures (Gauthier et al., 2005). Subsequently, the CD2 associated protein is essential for transferring VacA from early endosomes to late endosomes (Gauthier et al., 2005, 2007). The process of vacuolation is then dependent on syntaxin 7 and vesicle associated membrane protein 7 (VAMP7), both of which are integral to late endosomes and lysosomes (Suzuki et al., 2003; Mashima et al., 2008). Additionally, this process requires the vacuolar ATPase (V-ATPase) activity and dynamin, which are enzymes crucial for formation and stability of vesicles (Cover et al., 1993; Suzuki et al., 2001).
Induction of apoptosis
Although the fact that VacA causes apoptosis has been known for a while, the exact mechanism or mechanisms by which this occurs are still not completely understood. Evidence shows that VacA mediated apoptosis is dependent on interaction with the mitochondria (Kimura et al., 1999; Galmiche et al., 2000; Willhite et al., 2003; Willhite and Blanke, 2004; Oldani et al., 2009; Domanska et al., 2010; Foo et al.,2010). Indeed, VacA has been proven to reduce the membrane potential of the mitochondria, thereby allowing the release of cytochrome c (Kimura et al., 1999; Galmiche et al., 2000; Willhite et al., 2003; Willhite and Blanke, 2004). The modulation of the mitochondrial membrane potential by VacA also results in impaired cell cycle progression and a drop in ATP concentration (Kimura et al., 1999). Several early studies showed that VacA that is deficient in its ability to form channels inhibits cytochrome c release (Willhite et al., 2003; Willhite and Blanke, 2004), and blocks the ability to modulate the mitochondrial membrane potential (Willhite and Blanke, 2004), suggesting that channel formation is essential for these events.
However, some additional work has demonstrated that most VacA is localized to vacuoles inside host cells (Yamasaki et al., 2006). This finding suggests that VacA mediated cell death might not be a result of direct binding of VacA to the mitochondria, but perhaps suggests that VacA mediated induction of the pro-apoptotic factors in the Bcl-2 family might be involved. In keeping with this, it has been suggested that these pro-apoptotic factors actually interact with the mitochondria to release cytochrome c, and VacA has been shown to increase the level of pro-apoptotic Bax in a manner that mirrors the release of cytochrome c from the mitochondria (Yamasaki et al., 2006). Additionally, VacA can also induce the cleavage of poly (ADP-ribose) polymerase (PARP), by the activation of the death factor, caspase-3 in transfected cells. Furthermore, this cleavage can be inhibited by the overexpression of the pro-apoptotic factor Bcl2 (Galmiche et al., 2000; Yamasaki et al., 2006). Taken together, these data suggest that VacA has two potential mechanisms to induce apoptosis in intoxicated cells.
Disruption of cellular pathways
VacA deregulates multiple cellular pathways as well as inducing inflammation. VacA intoxication induces production of a variety of inflammatory cytokines that include TNFα, IL-1β, IL-6, IL-10, and IL-13 (Supajatura et al., 2002). Moreover, IL-8 is produced by several different cell lines in response to VacA mediated activation of the p38 MAPK through an increase in intracellular calcium and the subsequent activation of ATF-2, CREB, and NF-κB (Hisatsune et al., 2008).
VacA increases the activity of p38, ERK, and the activating transcription factor 2 (ATF-2; Nakayama et al.,2004). Through the p38/ATF-2 cascade, COX-2 is upregulated, which leads to increased production of prostaglandin E2 (PGE2; Hisatsune et al., 2007). Conversely, in mice VacA inhibits PGE2-stimulated duodenal epithelial bicarbonate (HCO−3) secretion by inducing the release of mucosal histamine (Tuo et al.,2009). While the reason for this discrepancy in VacA mediated increase in PGE2 effects is unclear, it should be noted that decreased duodenal epithelial HCO−3secretion is associated with duodenal ulcers and may leave the mucosal layer less able to repair itself (Isenberg et al., 1987). This could account for the role of VacA in gastric damage (Isenberg et al., 1987). Furthermore, VacA can also inhibit gastric acid secretion by increasing the mobilization of intracellular calcium, which in turn activates calpain 1 to hydrolyze the CagA targeted cytoskeletal protein, ezrin (Selbach et al., 2004; Wang et al., 2008). As well as the inflammatory pathway, VacA activates the p38 and the ERK pathways leading to deregulation of molecules that directly correlate with gastric damage.
Like CagA, VacA has also been shown to affect the β-catenin signaling pathway and therefore, perhaps the oncogenic potential of H. pylori. As stated earlier, deregulation of this molecule affects many cellular pathways involved in migration, cell cycle, polarity, and apoptosis, and numerous studies have demonstrated the effect that H. pylori has on the β-catenin pathway (Kurashima et al., 2008; Sokolova et al., 2008; Nagy et al., 2009; Nakayama et al., 2009; Tabassam et al., 2009). Recently, Tabassam et al. (2009) showed that VacA activates PI3K/p110α, which in turn activates Akt to phosphorylate GSK3β. This phosphorylation ultimately frees β-catenin to translocate to the nucleus to bind TCF/LEF and allows transcription of β-catenin-dependent genes such as cyclin D1 and potentially other oncogenic genes such as cdx1 (Lickert et al., 2000; Mizoshita et al., 2004; Tabassam et al., 2009). Thus this affect on the β-catenin pathway again lends credence to the fact that the more virulent alleles of VacA, which potentially could cause greater induction of this pathway, are associated with more severe disease manifestations.
Finally, VacA also affects the autophagy pathway; VacA induces the formation of autophagosomes and is associated with these structures (Terebiznik et al., 2009). Moreover, the stability of intracellular VacA is impacted by the presence of autophagosomes, and VacA stability is increased when autophagy is inhibited (Terebiznik et al., 2009).
Immune modulation
VacA has many roles, but one important function that may directly impact H. pylori colonization and persistence is its ability to act as an immune modulator. This immune modulation occurs through several distinct mechanisms. For instance, VacA disrupts the process of phagosome maturation through recruitment and retention of coronin 1, which is also known as tryptophan-aspartate-containing coat protein (TACO; Zheng and Jones, 2003). However, despite this disruption in phagosome maturation, VacA does not seem to impact the intracellular survival of H. pylori within monocytes (Rittig et al., 2003). Next, H. pylori infected macrophages form large vesicular compartments called megasomes and VacA supports this process by increasing homotypic phagosome fusion (Allen et al., 2000; Zheng and Jones, 2003). This allows H. pylorito persist in macrophages instead of being killed. VacA has also been proven to inhibit the invariant chain dependent pathway of antigen presentation by MHC class II molecules (Molinari et al., 1998b), and has been reported to interfere with the presentation of antigen in B cells (Molinari et al., 1998b). More recently, VacA has been reported to inhibit both PMA/anti-IgM and T cell induced B cell proliferation (Torres et al., 2007).
In addition to these pathways, T cells are also affected by VacA. VacA can enter activated, migrating primary T lymphocytes by binding to β2 integrin (CD18) and LFA-1 (Sewald et al., 2008b); LFA-1 is essential for this process since T cells deficient in LFA-1 are resistant to the effects of VacA (Sewald et al.,2008b). Intoxication by VacA can then inhibit the proliferation of CD8+ T cells (Torres et al., 2007) through down-regulation of the expression of the interleukin 2 (IL-2) surface receptor-α and inhibition of the production of IL-2, both of which are required for T cell proliferation and survival. These IL-2 effects occur through the inhibition of NFAT (Boncristiano et al., 2003; Gebert et al., 2003; Sundrud et al., 2004). This disruption in normal NFAT signaling may be due to blocked dephosphorylation of NFAT, which could occur by blocking the influx of calcium that is required for dephosphorylation by the calcium-calmodulin-dependent phosphatase calcineurin and subsequent nuclear translocation of NFAT (Boncristiano et al., 2003; Gebert et al., 2003). The down-regulation of IL-2 decreases cyclins D3 and E, which in turn decrease production of the retinoblastoma protein. This decrease induces cell cycle arrest in the G1 phase (Gebert et al., 2003).
With expression of over 100 genes altered in T cells upon intoxication with VacA (Gebert et al., 2003), it is perhaps not surprising that some redundancy exists in this process. A recent study found that VacA inhibition of CD4+ T cell proliferation is independent of NFAT induced IL-2 activation (Sundrud et al., 2004). Furthermore, VacA can induce the p38 MAPK pathway within T cells, neutrophils and macrophages (Boncristiano et al., 2003). In T cells, p38 is activated through activation of serine–threonine kinases (MKK3/6), which are linked to signaling molecules through a Rho family GTPase exchange factor, Vav (Bustelo, 2000; Ono and Han, 2000; Boncristiano et al., 2003). Through its exchange activity on Rac, Vav is linked to the reorganization of the cytoskeleton (Bustelo, 2000), and VacA uses Rac1 to rearrange the host cell cytoskeleton (Hotchin et al., 2000; Boncristiano et al., 2003). Taken together, all of these immune modulations by VacA probably allow for persistent infection with H. pylori. Additionally, perhaps the importance of immune modulation has led to the selective pressure to maintain expression of VacA in mostH. pylori strains.
Interactions Between CagA And VacA
There is a growing amount of literature that suggests that the CagA and VacA toxins interact, and that this interaction has an effect on disease severity (Jang et al., 2010). Early on, Yokoyama et al. (2005) showed an antagonistic effect between CagA and VacA on the NFAT pathway; CagA activates the NFAT pathway via activation of calcineurin through phospholipase Cγ, whereas VacA inhibits NFAT through prevention of calcineurin activation through decreased calcium influx due to VacA mediated pores (Yokoyama et al.,2005). Moreover, recent transfection assays showed that CagA blocks the apoptotic activity of VacA by two different mechanisms (Oldani et al., 2009); phosphorylated CagA blocks the ability of VacA to traffic to intracellular compartments, whereas unphosphorylated CagA blocks apoptosis in a manner that mimics Bcl2 (an anti-apoptotic factor) overexpression (Oldani et al., 2009). However, Bcl2 expression was not shown to be increased by CagA (Oldani et al., 2009). In fact, CagA not only blocks the cytotoxicity of VacA, but also blocks the ability of VacA to enter host cells (Akada et al., 2010).
Additionally, VacA and CagA show antagonistic activities in regards to cellular morphology. In cells co-cultured with isogenic H. pylori mutant strains deficient in cagA or vacA, increased vacuolation was seen in cells infected with cagA mutants, whereas cells infected with vacA mutants showed greater elongation of cells (Argent et al., 2008b). In other words, protrusion length was reduced in cells displaying vacuoles, and the number of vacuoles was decreased in elongated cells (Argent et al., 2008b). At a mechanistic level, activation of ERK1/2 by CagA is important for cell scattering and morphological changes (Neel et al., 2003), and VacA inhibits activation of ERK1/2 through inhibition of the activation of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor 2 (HER2/Neu; Tegtmeyer et al., 2009). One explanation for this antagonism, which was suggested by Akada et al. (2010) is that CagA is injected into the cells that the bacteria are attached to, which then protects those cells from the cytotoxic activity of VacA. VacA then proceeds to attack distant cells, thereby freeing nutrients. Overall, these combined interactions may explain our observation of a link between the most active VacA allele (s1/i1/m1), the most pathogenic CagA allele (EPIYA-ABD), and more severe disease manifestations (Jang et al., 2010).
Conclusion
Helicobacter pylori is a medically important bacterium that possesses a wide variety of virulence factors that allow it to thrive in the hostile environment of the stomach. The causal link between H. pylori infection and gastric cancer development has led to numerous studies designed to ascertain the role of virulence factors in the establishment of disease (Parsonnet et al., 1991, 1994; Talley et al., 1991; Blaser, 1998). Some virulence factors, such as HomB (Jung et al., 2009; Oleastro et al., 2009) and BabA (Gerhard et al., 1999; Mizushima et al., 2001) are just beginning to be linked via epidemiological evidence to support a role in the development of more severe disease. Conversely, CagA (Blaser et al., 1995; Kuipers et al., 1995; Xiang et al., 1995; Parsonnet et al., 1997; Huang et al., 2003) and VacA (Xiang et al., 1995; Letley et al., 2003; Rhead et al.,2007; Basso et al., 2008; Jang et al., 2010) have been studied extensively.
CagA was the first H. pylori virulence factor to be associated with more severe disease (Crabtree et al., 1991; Covacci et al., 1993; Blaser et al., 1995; Gwack et al., 2006) and has been shown to work on cellular processes that include β-catenin (Murata-Kamiya et al., 2007), ERK (Higashi et al., 2002b, 2004; Mimuro et al., 2002; Higuchi et al., 2004), and the inflammatory pathways (Brandt et al., 2005; Kim et al., 2006) to name a few (Figure (Figure1).1). This toxin works in both a phosphorylation dependent and independent manner, and polymorphisms located in the carboxyl terminus lead to differential induction of several cellular pathways (Higashi et al., 2002a,b; Hatakeyama, 2004). Since these polymorphisms affect the phosphorylation sites, one might assume that these variations would only affect pathways that are CagA phosphorylation dependent. However, even in its unphosphorylated state the sequence differences within this region of CagA affect the ability of the protein to multimerize, thereby leading to differential induction of CagA phosphorylation independent pathways as well (Kurashima et al., 2008; Lu et al., 2008). Meanwhile, a multitude of studies have assessed the effects of VacA on host cells (Figure (Figure2).2). This toxin also has a vast array of functions that span induction of apoptosis (Kimura et al., 1999; Galmiche et al., 2000; Willhite et al.,2003; Willhite and Blanke, 2004; Yamasaki et al., 2006) to modulation of the immune system (Figure (Figure2;2; Molinari et al., 1998b; Zheng and Jones, 2003; Torres et al., 2007). Again, there are polymorphisms within the VacA toxin that affect the range of cells it can intoxicate (Pagliaccia et al., 1998; Ji et al., 2000), as well as its ability to integrate into membranes and cause downstream effects (Letley and Atherton, 2000; McClain et al., 2001b; Rhead et al., 2007).
These two distinct toxins clearly have some overlap in their functions. Both are able to affect cell shape (Higashi et al., 2002b, 2004; Mimuro et al., 2002; Higuchi et al., 2004; Selbach et al., 2004; Wang et al.,2008), affect immune cells (Molinari et al., 1998b; Umehara et al., 2003; Zheng and Jones, 2003; Torres et al., 2007; Zhu et al., 2007), and activate oncogenic pathways such as β-catenin (Murata-Kamiya et al., 2007; Tabassam et al., 2009). They also clearly have antagonistic effects on each other, such as dampening the phenotypic effects on the host cell (cellular elongation induced by CagA vs. vacuolation caused by VacA; Argent et al., 2008b). CagA also has the ability to prevent VacA induced apoptosis, whereas VacA can prevent CagA induced nuclear translocation of NFAT (Yokoyama et al., 2005). It is believed that this antagonistic relationship exists to increase the life of the host cell (Argent et al., 2008b), and it has been shown that the more active form of VacA is often associated with the more active form of CagA and is thus, further linked to more severe gastric disease (Jang et al., 2010). As well as investigating the impacts of these toxins individually on host cells, more knowledge is needed on the interaction of these toxins and their combined impact on gastric disease.
Conflict of Interest Statement
The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or Department of Defense (DoD).
Acknowledgments
We thank Drs. Stephen Blanke and Richard Peek for critical reading of the manuscript. Research in the laboratory of D. Scott Merrell is supported by R01AI065529 from the NIH and R073PW from USUHS.
您还未取得发表评论权限,会员请登录.